Investigators are familiar with the institutional Human Research Protection Program, regulatory framework and ethical standards for protecting human subjects.
Human Research Question & Answer Guide
UK Human Research Protection Program
What rules or guidelines are you expected to follow?
Federal Regulations that Apply to All UK Human Subject Research
Department of Health and Human Services (DHHS) 45 CFR 46:
- Subpart A – “Common Rule” IRB Operations, Approval Criteria, Informed Consent
- Subpart B - Fetuses/Pregnant Women/Neonates
- Subpart C - Prisoners
- Subpart D - Children
Regulations that are applicable to select protocols:
- Food and Drug Administration regulations
- Health Insurance Portability Accountability Act (HIPAA), Family Educational Rights and Privacy Act (FERPA) or General Data Protection Regulation (GDPR)
Funding Agency Requirements:
- Department of Defense (DoD)
- US Department of Education (DoED)
- Environmental Protection Agency (EPA)
- US Department of Justice (DOJ); National Institute of Justice (NIJ); Bureau of Prisons (BOP)
- Department of Energy (DOE)
State Law
University of Kentucky Policies and Procedures and Regulations
President Level Administrative Regulations (AR):
- AR 7:1 Research Misconduct
- AR 7.2 Research Conflict of Interest and Financial Disclosure Policy
- AR 7:4 Human Research Subject Protection and Institutional Review Boards
- AR 7.9 Institutional Conflict of Interest
Vice President for Research (VPR): University of Kentucky Human Research Protection Program Comprehensive Plan
IRB/ORI:
- Standard Operating Procedures
- Application Forms
- IRB SURVIVAL HANDBOOK – Guidance Documents by Topic
What ethical standards or guides do you follow?
The above research regulations are based on the ethical principles set forth in the Nuremberg Code, Declaration of Helsinki, and the Belmont Report issued by the National Commission for the Protection of Human Subjects 1979. Belmont outlines three ethical principles that are central to human subject protection.
- Respect for persons involves recognition of the personal dignity and autonomy of individuals and special protection of those persons with diminished autonomy.
- Beneficence entails an obligation to protect persons from harm by maximizing anticipated benefits and minimizing possible risks of harm.
- Justice requires that the benefits and burdens of research be distributed fairly.
If you propose research be conducted at an international location, what do you inform the IRB regarding applicable local regulations, ethics review requirements, or cultural norms?
Identify Applicable Requirements/Protections
For research conducted at an international location, the investigator identifies local regulations, laws, or ethics review requirements for human subject protection. He/she may refer to the annual International Compilation of Human Research Standards or the National Institutes of Health ClinRegs website. If the project has been or will be reviewed by a local Ethics Committee or IRB, the investigator provides the UK IRB with a copy of that review.
Cultural Consultation
In addition, the investigator informs the IRB of any relevant cultural norms or customs particularly regarding recruitment or informed consent. The IRB obtains a cultural consultant to assist in the review of issues which require expertise beyond or in addition to that available on the IRBs. Cultural consultants provide comments, concerns, translations, in writing to the IRB on protocols involving non-English speaking subjects, and/or subjects from a foreign culture.
How does UK ensure the rights and welfare of participants are protected when the investigator is operating at a non-UK facility, in conducting collaborative research, or when oversight is shared with or deferred to another organization or IRB?
In the IRB application, investigators include letters of support approving the conduct of research at non-UK facilities.
If research involves collaboration with any sites and/or personnel outside UK, then it is considered multisite research and IRB reliance issues will need to be addressed.
UK has procedures to define the responsibilities of collaborating institutions and to coordinate communication among responsible IRBs. UK IRB requires a written agreement to be completed between organizations involved in a reliance relationship. A Reliance (or Authorization) Agreement identifies and describes the respective authorities, roles, responsibilities, and methods of communication between an institution/organization providing the IRB review of research and a participating site relying on the institution/organization.
Federal policies require review by a single IRB for select multi-site research. Studies using an external IRB MUST register with the UK IRB.
The ORI Single IRB Reliance website provides tools, checklists, forms, sample agreements and guidance for navigating single IRB review.
Prior to allowing investigators to cede research to an external IRB, the IRB (with assistance from ORI):
- verifies initial and continuing training of investigators regarding human subjects research. Information about ensuring initial and ongoing qualifications and Human Subjects Protection Training; and
- verifies the external IRB is accredited by the Association for the Accreditation of Human Research Protection Programs (AAHRPP). UK may agree to defer responsibility for IRB review to a non-AAHRPP accredited institution’s IRB for research that is not greater than minimal risk. To defer responsibility, the non-UK IRB must have an OHRP-registered IRB.
Does UK research have an emergency preparedness and response plan?
Yes, the Emergency Preparedness Response Plan provides guidance for initiating a response to an emergency/disaster situation impacting the UK Human Research Protection Program (HRPP) including plans to enable sustainability of the HRPP and continuing protection of participants in research during emergencies.
What Needs Institutional Review Board (IRB) Review?
Investigators understand the definition of human research and seek guidance when determining if an activity requires IRB review.
Could a Human Subject not have a heartbeat?
We aren't talking zombies, but the answer is yes.
Even though the regulatory definition of human subject specifies “living individual,” identifiable private information or identifiable biospecimens are also considered to be “Human Subjects.”
Under the definition of human subject, obtaining identifiable private information or identifiable specimens for research purposes constitutes human subjects research and requires IRB review.
"A Human Subject is a living individual about whom an investigator conducting research:
- Obtains information or biospecimens through intervention or interaction with the individual, and uses, studies, or analyzes the information or biospecimens; or
- Obtains uses studies, analyzes, or generates identifiable private information or identifiable biospecimens.” [45 CFR 46.102(e)]
Private information is considered a human subject if any study personnel or collaborators:
- can see identifiers;
- have access to a code linking identifiers;
- know who provided the private information; or
- can readily figure out who provided the private information.
For more guidance, see the brief video What Needs IRB Review? (5:10)
When does secondary research with specimens or data require IRB Review?
Activities that meet the federal regulatory definition of “research” with a “human subject” require IRB review. Secondary Research involves “re-using identifiable information and identifiable biospecimens that are collected for some other ‘primary’ or ‘initial’ activity.”
The following guidance provides a series of questions and a flow chart for determining whether secondary research requires IRB review. Considering only whether the data or specimens is “identifiable” may result in a wrong determination.
ORI also offers an interactive tool to point you to the most likely level of review based on responses to a series of questions.
What is the process for when determining whether an activity is under the purview of the IRB?
- See 'What Needs IRB Review?' for resources and contact ORI with questions on the application of regulations and UK policy
- If unclear, submit a Not Human Research (NHR) determination form on REDcap
- The ORI Director or IRB Chair provides an official determination on whether an activity meets applicable regulatory definitions of human subject research
- The ORI communicates the decision to the investigator via phone, email, or hard copy memo
IRB Submission & Review Types
Investigators are familiar with the different types of IRB review and the basics of the submission process.
How do I find out general information about the UK IRB and human research?
The IRB FAQs answer common questions regarding review operations, Medical vs. Nonmedical IRB, informed consent, terminology, etc. Categories include General IRB, Need for IRB Review, Informed Consent, and Continuing/Annual Administrative Review.
A printable FAQ handout provides answers to select questions for researchers or faculty new to the UK IRB process.
Where should I start to determine what type of IRB review will be required?
ORI has a Getting Started webpage for individuals new to human subject research. The ORI IRB Review Types webpage provides resources for determining which type of review a protocol will require.
Exempt and Expedited review is designed for minimal risk research. However, each category has conditions and limitations. Research that cannot meet the criteria for exempt or expedited review must be submitted for full review by a convened board.
In addition to checking the ORI guidance, researchers are encouraged to contact ORI for consultation and a preliminary interpretation of the most appropriate application type. Ultimately the IRB will choose the type of review based on the full application relative to the regulatory and ethical framework.
| Review Type | Risk/Initial Review Categories | Initial Review by: | Guidance |
|---|---|---|---|
| Exempt | Minimal Risk/6 Categories | 1 IRB member (consultant if necessary) | UK ORI New Common Rule Exemption Categories Tool |
| Expedited | Minimal Risk/7 Categories | 1 IRB member (consultant if necessary) | Issues to be Addressed when conducting Expedited Review |
| Convened | Greater than Minimal Risk or doesn't fit in above categories | Full Board Meeting with Investigator Invited |
Research Risk Assessment Guidance |
How do I request IRB approval for changes while conducting the research?
Modification Requests (MR) – submit an MR for any change to a protocol from what was previously approved. This includes proposed changes to the current IRB approved protocol or changes which impact an individual subject but does not change the overall protocol (i.e., Exception or Deviation)
- Exception - one-time enrollment of a research subject in a protocol that fails to meet current IRB approved
- Deviation - one-time departure from the current IRB approved protocol once a subject has been enrolled
*Changes may not be initiated without IRB review and approval, except where necessary to eliminate immediate hazard.
Continuing Review (CR) or Annual Administrative Review (AAR)– changes may also be requested as part of the annual or continuing review submission.
Does ORI provide educational resources for researchers?
Yes, In addition to resource websites such as Getting Started and the IRB Survival Handbook, the ORI Education Team has built an Education Resource library of topic-specific videos, recordings, and interactive tools. These resources aim to provide clear and accurate content, in a brief and engaging format.
What information or resources are available for conducting survey research?
The ORI Survey Website includes survey guidance and tools developed to meet the diverse needs of survey researchers. It includes identifiable and anonymous consent templates, resources for advertising, survey platforms, fraud prevention, and regulations applicable to school-based surveys.
Scientific Design & Minimizing Risk
In addition to researchers, who is involved in conducting scientific review of human research at UK?
- Department Chairperson/Faculty Advisor attests in the IRB application, that the science is meritorious and deserving of conduct in humans by considering the:
- validity and utility of science;
- availability and qualifications of personnel;
- potential subject population; facilities and equipment;
- ongoing mentoring and guidance; and
- resolves issues prior to the IRB’s receipt of the submission.
- The IRB considers the scientific study design only within context of human subject protection and risk benefit analysis. IRB members draw on their own knowledge and disciplinary expertise to determine if research procedures are consistent with sound research design and the protocol has potential to yield the expected knowledge. When needed, the IRB seeks consultation from content experts.
What is the minimum IRB requirement for maintenance of research records?
At a minimum, research records should be maintained for six (6) years after completion of the study. Longer retention may be required by sponsors or for studies that fall under the authority of other agencies. For more information see the ORI/IRB Study Closure SOP.
Note: this IRB Record Retention requirement is separate from the University’s Data Retention and Ownership Policy which applies to all types of research.
What additional information privacy regulations apply to select protocols?
- Health Insurance Portability and Accountability Act (HIPAA) is a federal regulation designed to protect the use and disclosure of Protected Health Information or PHI. PHI is defined as any of the 18 HIPAA identifiers in combination with health information transmitted or maintained in any form (electronic, paper, or oral) that relates to the past, present or future physical or mental health or conditions of an individual.
- Family Educational Rights and Privacy Act (FERPA) is a federal law that protects the privacy of personally identifiable information contained within a student’s educational record.
- General Data Protection Regulation (GDPR) is a regulation affecting the way data is processed in the European Economic Area (EEA). This regulation increases the rights afforded to research participants and reshapes the way organizations handle and process personal data from individuals located in the EEA.
Click each title for information or guidance on when and how these impact human research.
What is the difference between protecting the privacy interests of participants and maintaining the confidentiality of data?
Privacy concerns people.
The following are considerations and strategies for respecting the privacy of potential participants:
- Consider the methods used or setting where potential participants are identified. What is the targeted study population's expectation of privacy, both in person and online?
- Only approach individuals known to you or make contact on behalf of someone the individual knows.
- Comply with privacy guidelines of applicable professional associations and scholarly disciplines (e.g., oral history, anthropology, psychology).
- Access the minimum amount of information necessary.
Confidentiality concerns data.
Confidentiality refers to the researcher’s agreement with the participant about how the participant’s identifiable private information will be handled, managed, and disseminated. In the IRB application, investigators describe their plan to preserve the confidentiality of identifiable data, including:
- controls on storage, handling, and sharing of data;
- physical security measures (e.g., locked facility, limited access);
- data security (e.g., password-protection, data encryption) see IRB Data Security Guidance;
- safeguards to protect identifiable research information (e.g., coding, certificate of confidentiality);
- procedures employed when sharing material or data, (e.g., honest broker (if applicable), written agreement with recipient not to re-identify); and
- measures that you will take to secure and safeguard confidentiality if protocol involves storing or sharing information or tissue/specimens/data for use in current or future research.
How do IRB regulations define minimal risk?
The Department of Health and Human Services defines minimal risk to mean “the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” [45 CFR 46.102(2)(i)].
Research risks may be categorized as physical, psychological, sociological, economic, and legal.
How does the IRB assess the risk-benefit ratio of the research?
In order to approve human research, the IRB must determine that potential risks to subjects are reasonable in relation to anticipated benefits to subjects, science and society.
How might investigators minimize risks in research?
- Using procedures already being conducted for non-research reasons
- Incorporating criteria to exclude “at risk” subjects
- Choosing least intrusive design that yields valid data (outcomes vs. randomized intervention; comparative drug vs. placebo)
- Conducting safety monitoring including safety labs and other assessments
- Planning for responding to clinically significant abnormalities including withdraw of study product and re-challenge with product if appropriate
- Including provisions for medical services or professional intervention (e.g., counseling) in the event of adverse events
- Adopting strategies for research with a focus on, treatment for, or potential for suicidal ideation or behaviors. See the ORI Guidance on Suicidality and Research Ethics
- Ensuring protections to secure confidential or private identifiable information
- Establishing data and safety monitoring
- Obtaining a Certificate of Confidentiality to protect against compulsory legal demands such as subpoena
Conflict of Interest (COI)
Investigators and research staff should understand the organization’s conflict of interest policy in order to follow it. For example, investigators should know what interests the organization requires to be disclosed. Investigators and research staff should know how, when, and to whom to disclose interests.
What is UK's policy on Conflict of Interest (COI)?
UK actually has two policies on conflict of interest; one for research investigators and one for the institution itself.
Financial COI related to research of individual investigators is covered in Administrative Regulation (AR) 7.2 - Financial Conflicts of Interest in Research. The AR outlines procedures for defining, identifying, disclosing, managing, reporting and training regarding COI.
A potential or actual Conflict of Interest (COI) exists when a significant financial interest (as defined below) of an Investigator or a family member of the Investigator could directly and significantly affect the design, conduct, or reporting of research.
COI is administered by the Office of Sponsored Projects Administration (OSPA).
See the OSPA COI website for guidance.
If you have questions or need assistance with a specific situation, contact Conflict of Interest Administrator Emily Bradford at 257-9420 or emily.bradford@uky.edu.
ORI & OSPA coordinate handling of Investigator COI for both funded and unfunded human subject research.
How is researcher COI managed?
The IRB application asks if any investigators or key study personnel have a Significant Financial Interests requiring disclosure and if the interests are related to the proposed research?

- If a financial COI exists and cannot be eliminated, the investigator works with Emily Bradford to complete the University of Kentucky template management plan and obtains approval from their Associate Dean for Research (ADR).
- All management plans are referred to the Research Conflict of Interest Committee (RCOIC) for review.
- The RCOIC recommends a plan which is submitted to the Vice President for Research (VPR) for approval.
- The IRB does not complete its review and approval of the IRB application until it receives the final VPR-approved management plan. The IRB may not change the approved plan, but it may impose additional restrictions/conditions on the protocol (e.g., disclose conflict in the informed consent document).
Monitoring & Reporting Requirements
Investigators assess and report unanticipated problems occurring during a research study in accordance with applicable federal, state, and local regulations and the Organization's policies and procedures.
What are examples of events/issues that you report to the IRB?
The Investigator Quick Guide is a one-page handout listing categories of events/issues to report including unanticipated problems, violations, unresolved subject complaints, federal agency inspections, data and safety reports, subject incarceration, etc.
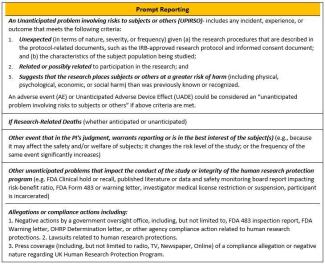
What unanticipated problems or adverse events are investigators required to promptly report to the IRB?
The UK IRB Policy on Unanticipated Problems and Safety Reporting requires investigators to promptly report Unanticipated Problems Involving Risks to Subjects or Others (UPIRSO) as well as other safety-related information important to human subject protection or study integrity. See the policy for reporting timeframe requirements for each category. The Prompt/NonPrompt Reporting guide was developed to help research teams understand the differences between an internal prompt event and an internal non-prompt event, based on the first three prompt reporting criteria in the UK UP Policy.
Qualifications, Training, & Oversight
Investigators and research staff are qualified by training and experience for their research roles, including knowledge of applicable federal, state, and local regulations; relevant professional standards; and the Organization's policies and procedures.
Investigators appropriately delegate tasks that are commensurate with staff qualifications and provide oversight throughout the study.
What Responsibilities and Qualifications are required for research investigators?
The Principal Investigator’s Guide to Responsibilities, Qualifications, Records and Documentation of Human Research list responsibilities and basic qualifications for conducting human subject research.
It is a succinct guide to enable compliance and includes an investigator’s local UK responsibilities when research reviewed by an external, Non-UK IRB.


What are the IRB's expectations regarding oversight and delegation for medical intervention studies and clinical trials?
Unless a study submitted to the Medical IRB is non-interventional, (e.g., survey, record review, or purely outcomes research), some form of medical oversight may be necessary. However, the degree and level of expertise needed can vary depending on risk level, condition, study population, applicable regulations, and external oversight (e.g., sponsor monitor; mentor). The Investigator Qualifications and Provision of Medical Oversight guide provides regulatory guidance on oversight, supervision, and delegation.
Inappropriate delegation is frequently cited in FDA warning letters. The FDA Guidance, Investigator Responsibilities — Protecting the Rights, Safety, and Welfare of Study Subjects clarifies FDA’s expectations on delegation and oversight.
- The Investigator Qualifications and Provision of Medical Oversight guide provides regulatory guidance on oversight, supervision, and delegation.
What is your process for task delegation?
Prior to describing delegation, the Principal Investigator should indicate his/her direct involvement in the conduct of the study including recruitment, obtaining consent, assessing eligibility criteria, events, and protocol procedures.
An investigator may delegate many tasks to study staff, if the tasks is within their scope of practice. Anyone delegated a task, must be qualified by education, training, and experience (and state licensure where relevant). In addition, the investigator would need to ensure that the delegation is consistent with any specifications in the research protocol or stipulations by the IRB. Medical procedures and assessments (including adverse event causality, un-blinding, treatment decisions) should not be inappropriately delegated to unqualified staff. Using task delegation logs is a best practice for clinical research, to define roles and indicate who serves in the PI’s absence.
Investigators designate which study personnel should be authorized to obtain informed consent on the IRB application for review by the IRB. The investigator must assure that the study personnel are informed regarding their obligations and commitments.
Data & Safety Monitoring
The Investigator designs and carries out research studies with adequate data and safety monitoring during the research, when appropriate.
Which Studies require a Data and Safety Monitoring Plan (DSMP) at UK?
A DSMP is required at Initial IRB Review for:
- Greater than minimal risk research
- NIH Funded Clinical Trial
- FDA-Regulated Clinical Investigation
Monitoring the progress of the research and the safety of participants are key components to a DSMP.
The IRB recognizes that the elements of a monitoring plan may vary depending on the potential risks, complexity, and nature of the research. After reviewing the plan, the IRB may determine that a formal DSMP is not necessary or find that monitoring by the investigator as proposed is adequate or could determine that the study requires an independent individual or independent body (i.e., Data and Safety Monitoring Board) for monitoring.
The ORI DSMP website provides guidance for developing a plan and guidance for when a Data and Safety Monitoring Board (DSMB) is warranted or required. NOTE: If relying on an independent agent or committee for DSMB services, (e.g., Center for Clinical and Translational Science DSMB), it is the PI’s responsibility to establish the services with the agent or committee.
What Data and Safety Monitoring information are you required to report to the IRB?
Reporting status and outcomes from Data and Safety Monitoring is essential for the IRB to decide if the criteria for approval is met at continuing review. The Principal Investigator (PI) is responsible for reporting on the status of the studies’ data and safety monitoring plan. ORI staff contact the PI at Continuing Review (CR), if a report is expected and not submitted. If the study has an independent Data and Safety Monitoring Board (DSMB) the PI should submit activity reports to the IRB upon receipt, through modification requests or continuing review submission.
Recruitment & Study Population
Investigators employ fair and equitable recruitment and avoid undue influence or coercion.
Investigators should have a justifiable rationale for inclusion of vulnerable populations.
If vulnerable populations are to be recruited, investigators comply with regulatory requirements and apply additional safeguards for protecting the subjects' rights and welfare.
Describe some key points for ethical and respectful recruitment methods.
Respect Privacy – consider time and setting for recruitment activities, including online settings
Equitable Selection - Proportionate Distribution; not targeting or excluding based on convenience
Undue Influence – No finders fees or recruitment bonus to study staff; appropriate IRB-approved ads; use of third party if investigator is an authority figure
No Cold Contacts – contact by personnel with legitimate access or through individuals with established relationships
Compensation – appropriate amount, method, and timing
What additional provisions do you employ for protection of vulnerable populations, groups vulnerable to undue influence, or populations with cultural considerations?
There are additional provisions for protection of vulnerable populations and potential participants who are vulnerable to coercion or undue influence.
See the following Guidance/Policy Documents for groups included in your research:
- Adults with Impaired Consent Capacity Policy
- UK IRB Policy on Children in Research
- Protection of Human Subjects in Research Involving HIV Testing
- Summary on Prisoners Regulations – OHRP Video Series Prisoner Review
- Guidance for Enrolling Students as Research Subjects
- Guidance for Enrolling K-12 Students as Research Subjects
- Research Involving Economically or Educationally Disadvantaged Persons
Informed Consent Process & Documentation
The Investigator develops an informed consent process appropriate to the research and population emphasizing comprehension and voluntary participation. Investigators understand the difference between consent process (which is ongoing) and consent documentation.
The ORI Informed Consent website serves as a one-stop website with informed consent resources ranging from creation to documentation and everything in between. Linked pages include guidance on various processes, situations, and subject populations. Also, includes links to externally launched external consent trainings.
Describe your informed consent process?
- The informed consent process involves the “who”, “what”, “when’, “where”, and “how” that result in a valid, effective informed consent. Investigators indicate in the IRB application, which study personnel will obtain consent and describe the proposed process in the research description.
- In developing a robust process, consider techniques to ensure comprehension and voluntary participation, such as:
- plain language documents; visuals, graphics;
- steps to minimize coercion or undue influence; &
- teach back questions to assess understanding.
- Potential participants must be allowed ample time to read, review, discuss and consider participation.
- While the informed consent process is prospective and takes place prior to any research activity. Consent should also be an ongoing educational interaction between the investigator and the research participants that continues throughout the study. Enrolled participants must be provided information arising during the study, which may affect their willingness to continue to participate.
What is the difference between Informed Consent, and the process of obtaining Assent and Parental Permission?
Because children and some adults with impaired consent capacity are not legally considered capable of providing consent, regulations do allow a parent or legally authorized representative (LAR) to give “permission” for the individual to participate when assent or “affirmatively agreement” to participate is obtained from the child (or adult with impaired consent capacity). Depending on the risk level of the study, provisions may be necessary for permission of both parents.
- For guidelines on when assent needs to be documented based on age and maturity, as well as parental permission requirements for research involving children, see the UK IRB Policy on Children in Research.
- Refer to the UK Impaired Consent Capacity Policy for guidelines on developing a plan that evaluates level of impairment given the context of the research. Researchers can respect even limited autonomy by obtaining participant assent and recognizing that individuals are always considered competent to refuse.
What is Informed Consent Key Information?
One of the new regulatory requirements from the Revised Common Rule states that:
“Informed consent must begin with a concise and focused presentation of the Key Information that is most likely to assist a prospective subject in understanding the reasons why one might or might not want to participate.” [45CFR46.116(a)(5)(i)]
Key Information is not defined in the Common Rule, but the regulation indicates that it should be brief, presented first, and zero-in on the key pros and cons a reasonable person would weigh rather than having that information buried deep in a detailed consent document.
It is generally NOT inclusion or exclusion criteria except when the criteria affect choice. For instance, if a key factor involves criteria that may be undesirable to the reasonable person (e.g., “you might not want to participate if you would be unable or unwilling to abstain from alcohol for the next 30 days.”).
Key Information Samples for Simulated Studies are available on the Sample Applications and Protocol Development Resources webpage.
Does the Key Information need to be a full page if the entire consent is not much longer than a page?
The regulation is not prescriptive or rigid about the format or content of Key Information. The intent is to allow consent documents (and the process) to be tailored to the particular research. The Preamble to the Revised Common Rule acknowledges that, for some relatively simple studies, the entire document will be brief. In such cases, the key information may fit within a paragraph vs. an entire page given the context of the study.
Under what condition can informed consent be altered or waived for minimal risk research?
Some research projects would not be possible if informed consent were required. The IRB may approve a consent procedure that does not include, or which alters, some or all of the elements of informed consent, or may waive the requirements to obtain informed consent if it finds and documents that the research meets the Common Rule Regulatory conditions:
- The research involves no more than minimal risk to the subject.
- The rights and welfare of subjects will not be adversely affected.
- The research could not practicably be carried out without the requested waiver or alteration.
- Whenever possible, the subjects or legally authorized representatives will be provided with additional pertinent information after they have participated in the study.
- If the research involves using or accessing identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format. Private information/specimens are “identifiable” if the investigator may ascertain the identity of the subject or if identifiers are associated with the information (e.g., medical records). This could be any of the 18 HIPAA identifiers including dates of service.
NOTE: FDA also allows waiver of informed consent for FDA-regulated minimal risk research. Where HIPAA applies to the research, a HIPAA waiver of Authorization may also be required.
Under what circumstances can documentation of informed consent be waived?
IRB regulations allow the IRB to waive the requirement to obtain a signed consent document for some or all the subjects if certain conditions are met.
The IRB waives the signature requirement but may require the investigator to provide participants with a document or written statement regarding the research.
The following are options for waiving documentation:
- The only record linking the subject and the research would be the consent document and the principal risk would be harm resulting from breach of confidentiality. Under this option, each participant (or legally authorized representative) must be asked whether (s)he wants to sign a consent document; if the participant agrees to sign a consent document, only an IRB approved version should be used.
- The research presents no more than minimal risk and involves no procedures for which written consent is normally required (i.e., a cover letter on a mailed survey or phone script)
- Subject or LAR is a member of a distinct cultural group or community in which signing forms is not the norm. The research is no greater than minimal risk and there is an alternative mechanism for documenting that informed consent was obtained.
NOTE: HIPAA regulations do not have an equivalent waiver of documentation of HIPAA Authorization. FDA currently allows waiver of documentation only under Option 2 [21CFR56.109(c)].
Complaints, Concerns, Suggestions, Questions or Requests for Information
Investigators respond to participant complaints or requests for information.
Who do you contact with a complain, concern, or suggestions?
Concern regarding ORI and IRB administrative procedures -
ORI Director Helene Lake-Bullock
(859) 257-2978 or
helene.lake-bullock@uky.edu
Concern regarding an IRB Decision:
- Contact the applicable IRB Chair
- Submit a written appeal that includes a justification for changing the IRB decision. The convened IRB reviews the appeal. The appeal determination final.
What provisions do you have in place for receiving and handling a subject complaint of request for information?
- Your protocol-specific plan described in the IRB Research Description.
- The procedures to satisfy this should offer a safe, confidential, and reliable channel for current, prospective, or past research subjects (or their designated representative) allowing them to discuss problems, concerns and questions, or obtain information.
- For greater than minimal risks studies, the IRB recommends the consent document(s) include a reliable, dedicated pager or phone number for after-hours emergencies.
Who may a subject call, outside of the study personnel, about their rights and welfare?
Each IRB approved informed consent document should include the ORI toll-free phone number (1-866-400-9428) as a subject's primary contact point about their rights and welfare.
Food & Drug Administration (FDA) Regulated Research
Investigators are responsible for ensuring that studies testing FDA-regulated products are conducted under a valid Investigational New Drug (IND), Investigational New Device (IDE), meets Abbreviated IDE requirements or is exempt from IND/IDE requirements.
Investigators are responsible for the control and accountability of FDA-regulated investigational products.
Investigators follow FDA regulations and UK procedures for emergency use of a test article.
What is the difference between Single Subject Emergency Use and Planned Emergency Research?
The single subject emergency use provision in the FDA regulations [21 CFR 56.104(c)] is an exemption from prior review and approval by the IRB for an investigational drug or device to be used in a human in a life-threatening situation where time is not sufficient to obtain IRB approval. FDA regulations require that any subsequent use of the investigational product at the institution have prospective IRB review and approval.
FDA regulations for planned emergency research [21 CFR Subpart B 50.24] provide a narrow exception to the requirement that the investigator obtain informed consent from each subject, or the subject's legally authorized representative, prior to enrollment in research conducted in an emergency setting. The regulations also provide additional protections for subjects enrolled in these Exception from Informed Consent (EFIC) studies.
For example, the regulations require advance public disclosure and consultation with representatives of the communities in which the clinical investigation will be conducted. They also require public disclosure of enough information following completion of the clinical investigation to apprise the community and researchers of the study. As well, the regulations require that an independent data monitoring committee exercises oversight of the clinical investigation.
What resources are available for investigators conducting FDA-regulated research?
The ORI FDA Research Question and Answer website provides training materials, interactive tools, resources, and information on multiple areas of FDA-regulated research including research testing drugs, devices, software as medical device, and treatments including emergency use, expanded access, and Humanitarian Use Devices.
Community Engaged Research (CER)/Community Based Participatory Research (CBPR)
Resources to facilitate CER/CBPR
What resources are available to investigators conducting Community Engaged Research (CER) or Community Based Participatory Research (CBPR)?
ORI provides a CBPR frequently asked questions (FAQs) guide to assist researchers in the design and implementation of research in the community and facilitate Institutional Review Boards’ review of CER/CBPR.
CBPR investigators may work with ORI education, to propose use of non-traditional, CBPR-focused human subject protection training for community partners. The CBPR FAQs provides links to several national programs available designed to educate nonscientific/nonresearch community-based individuals. In addition, UK ORI has developed K-12 Teacher & Support Staff Human Subject Protection (HSP) Training specifically designed to educate teachers and support staff on protecting the rights and welfare of students.
What are considerations for IRB review of CBPR?
- How research involves community members or partners in the design and implementation of the study!
- Community risks, benefits, and consent.
- How results will be shared with the community.
- How protocols may be developed to include flexibility (e.g., range of procedures; semi-structured interviews within defined parameters).
Outreach & Education for the Public and Potential Research Participants
Investigators are aware of public education and potential research participant outreach efforts.
Who provides outreach to educate the public and potential participants about human subject research?
The Participant Recruitment/Marketing core of the Center for Clinical and Translational Science (CCTS) works with UK Healthcare, UK Marketing, ORI, and research investigators to provide education, outreach, and research opportunities to the public. The CCTS Participant Website provides several mechanisms for the public to learn about research participation including videos, kiosks, and databases.
The ORI Participant Website provides additional participant education links as well as contact information for subject concerns, suggestions or questions.
Feasibility & Resources
Investigators assess feasibility and ensure adequate resources to perform research.
How do you assess and ensure availability of resources required to conduct research in a way that will protect the rights and welfare of participants?
- Protocol considerations include valid research question, risk vs. potential benefit, realistic inclusion/exclusion criteria, appropriate facilities, sufficient time, appropriate staff credential or expertise, adequate potential subject population, safety considerations such as placebo or washout, etc., personnel, space, equipment, and time.
- Facility Considerations - consider proximity or availability of other resources. For example, the proximity of an emergency facility for care of participant injury, or availability of psychological support after participation. Investigators should not commence a research study without adequate resources to protect participants and should stop a research study if resources become unavailable.
- Potential Subject Population – the CCTS provides biostatistics and informatic consultation, as well as a query tool (i2b2) for searching clinical data based on inclusion criteria to determine adequacy of a potential patient population. CCTS Participant Recruitment Services a assist in developing recruitment plan, creating recruitment materials, and promoting IRB-approved studies.